min. read
Topics
Researchers discover how mitochondrial fusion regulates neuromuscular junction synapse development
Image
Mitochondria are double membrane cytoplasmic structures that act as cellular powerhouses by generating ATP. Neurons are metabolically very active and have high energy requirements. Also, unlike other cell types, they have long processes and it is not unusual for axons of certain neurons to project many meters out of the main cell body (soma). Therefore, regulating mitochondrial trafficking and dynamics are critical for optimal functioning of neurons.
While it is known that defects in mitochondrial transport or dynamics in neurons result in neurological or neurodegenerative diseases, it is not clear what the underlying causative steps are. For instance, while it was known that loss of mitochondrial fusion protein, Mitofusin 2 (Mfn2) causes Charcot Marie Tooth type 2A (CMT2A), a progressive and severe loss of motor neurons, the mechanism by which functional loss of Mfn2 result in neuropathy is not clear.
A recent paper from Dr. Hugo Bellen’s lab at Baylor College of Medicine and Neurological Research Institute at Texas Children’s Hospital provides a clue. This study has identified a critical role for mitochondrial fusion in the development of synaptic junctions by regulating the synthesis of a steroid hormone.
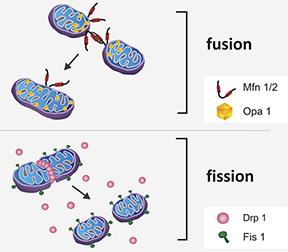
Mitochondria undergo repeated cycles of fusion and fission that helps to create new mitochondria as well as maintain genetic integrity of the existing mitochondria. Mitochondrial fusion is a two-step process in which outer membranes of two nearby mitochondria are tethered and brought closer together by Mfn 1 and Mfn2 followed by Optic atrophy protein (Opa1)-mediated fusion of inner membranes. A dynamin-like protein (Drp1) is recruited to the outer mitochondrial membrane during mitochondrial fission to constrict and sequentially separate mitochondrial inner and outer membranes.
Image
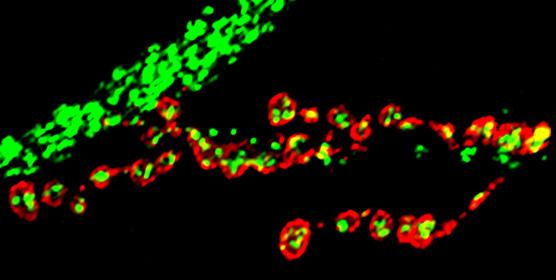
In the study published in Elife, researchers from Dr. Bellen’s group found that larvae of mitochondrial fusion mutants (Marf) had smaller, rounder as well as fewer mitochondria at the nerve terminals, consistent with the known functions of mitofusins in mitochondrial fusion and axonal transport respectively. Not surprisingly, these neurons generated less energy at the terminals and were unable to properly signal to their targets. Interestingly, all these defects were overcome when Marf protein was added back to the motor neurons.
The most striking and novel observation of this study was that unlike previously studied fission mutants, mitochondrial fusion mutants, Marf and Opa1, displayed several defects in neuromuscular junction development. Surprisingly, they found that this could not be attributed to aberrant Marf or Opa1 function in neurons, muscles or glial cells but was rather due to their dysfunction in distant, hormone-producing tissues called the ring glands.
Ring glands are sites where steroid hormones essential for growth, differentiation of various tissues as well as larval molting and metamorphosis are produced. These steroid hormones are synthesized from cholesterol via pathways that are evolutionarily conserved from flies to mammals.
Interestingly, researchers found while both outer and inner mitochondrial fusion proteins were required for steroid hormone synthesis, only outer mitochondrial fusion proteins are sufficient for certain key intermediate steps such as lipid droplet synthesis and cholesterol ester storage. It is important to note that disruption of mitochondrial fission did not have any effect on hormone synthesis.
Moreover, they found that hormone production in these mutant flies could only be restored if both vertebrate mitofusins (MFN1 and MFN2) were added back together. This indicates that the ancestral fruit fly Marf has functionally diverged to result in two vertebrate mitofusins that have non-overlapping yet complementary functions.
In summary, this study shows that improper mitochondrial fusion results in aberrant hormone synthesis and defective synapse development, which may be a potential mechanism underlying CMT2A neuropathy. Although this hypothesis remains to be validated in vertebrate rodent models and CMT patients, this study highlights the importance of using fruit flies as a genetic tool to quickly unravel complex mechanisms involved in the pathogenesis of various diseases.
Related Stories





20731
Press Release
article