min. read
Topics
Loss of Frataxin causes neurodegeneration due to iron toxicity
Image
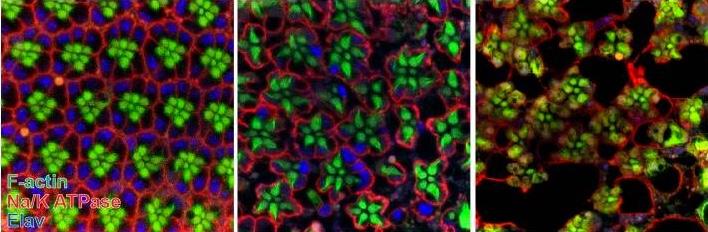
A recent study published in the journal eLife by Dr. Hugo J. Bellen’s team at Baylor College of Medicine and Neurological Research Institute at Texas Children’s Hospital has provided the first experimental evidence that loss of frataxin may cause neurodegeneration via iron toxicity.
Fredreich’s ataxia (FA) is a progressive neurodegenerative disorder. Most individuals have onset of FA between the ages of 5 and 18 years. <25% of the diagnosed individuals have adult or late onset FA, which can occur anytime during adulthood. The most common symptoms include loss of coordination, fatigue, vision/hearing impairment, slurred speech, diabetes and heart disease, although their mental capabilities remain intact.
FA is caused by mutations in Frataxin (FXN) gene. FA is inherited in an autosomal recessive manner, meaning that individuals must have two mutated or abnormal copies of this gene. This happens when both biological parents carry at least one copy of the mutated gene and contribute that defective copy to the offspring.
FXN is responsible for proper assembly of iron-sulfur clusters that are incorporated into many other metalloproteins, including proteins critical for mitochondrial function. Loss of FXN disrupts the electron transport chain (ETC), a series of oxidative and reductive reactions that produce cellular energy. Impaired ETC results in massive production of chemically unstable reactive oxygen species (ROS) that are toxic to the cells. Until recently, it was proposed that in FA patients, loss of FXN function damaged neurons due to excessive production of ROS.
However, contrary to this hypothesis, loss of FXN lead to only a modest increase in ROS levels in several animal models. Also, FA patients undergoing anti-oxidant therapy have shown limited improvement in many clinical trials.
In this study, researchers found that fruit flies lacking frataxin had excessive iron accumulation in the neurons, which was accompanied with a age-dependent neurodegeneration of eye receptors. Interestingly, while mitochondrial function was perturbed in these neurons, there was no significant increase in ROS levels.
Instead, they observed an increased synthesis of sphingolipids, a class of fatty acids that are commonly found in neurons and are involved in cellular signaling. Consistent with this observation, a previous study from a different laboratory had demonstrated that cellular iron toxicity is mediated through sphingolipid signaling.
Interestingly, increased sphingolipid signaling was accompanied by an unusual activation of two other proteins, 3-phosphoinositide dependent protein kinase-1 (Pdk1) and myocyte enhancer factor-2 (Mef2). Decreasing iron toxicity, inhibiting sphingoside formation or reducing the levels of Pdk1 or Mef2 effectively suppressed neurodegeneration in these mutants.
This study provides a first glimpse into a novel causative mechanism for FA. If validated in humans, this could be prove to be a very important step towards developing an effective therapy for FA patients.
Related Stories




20611
Press Release
article